
AceFF Machine Learning Potentials
Collection
The best machine learning force-field for drug discovery
•
3 items
•
Updated
•
1
Organization(s): Acellera Therapeutics, inc
Contact: info@acellera.com
License: apache 2.0
Acellera AceFF 2.0 is a next-generation machine learning interatomic potential (MLIP) designed for small molecules. It addresses key limitations of traditional molecular mechanics (MM) force fields and earlier NNP models, including restricted atom types, limited charge support, and computational inefficiencies.
The model leverages the TensorNet v2 architecture [1] and the NNP software library TorchMD-Net [2] to provide accurate predictions for diverse drug-like compounds, supporting all key chemical elements and charged molecules. Acellera AceFF 2.0 improves the stability of molecular dynamics simulations, supports 2 fs timesteps, and achieves state-of-the-art accuracy with fewer outliers in RBFE predictions.
Acellera AceFF 2.0 is the second version of a new family of potentials released by Acellera. It uses TensorNet 2-layers trained on Acellera's internal proprietary dataset of molecular forces and energies using the wB97M-V/def2-tzvppd level of theory and VV10 dispersion corrections.
The training set was built on PubChem. We extracted the SMILES and generated molecules, filtering out molecules larger than 30 atoms. We kept only molecules with the elements H, B, C, N, O, F, Si, P, S, Cl, Br, and I.
The table shows the results on the Wiggle150 benchmark. We include AIMNet2 and ANI-2x for comparison.
| Method | MAE (kcal/mol) | RMSE (kcal/mol) |
|---|---|---|
| AceFF-2.0 | ||
| AceFF-1.1 | 2.51 | 3.18 |
| AceFF-1.0 | 2.73 | 3.32 |
| AIMNet2 | 2.39 | 3.13 |
| ANI-2X | 4.41 | 5.41 |
Performance of MLIPs on Wiggle150 benchmark
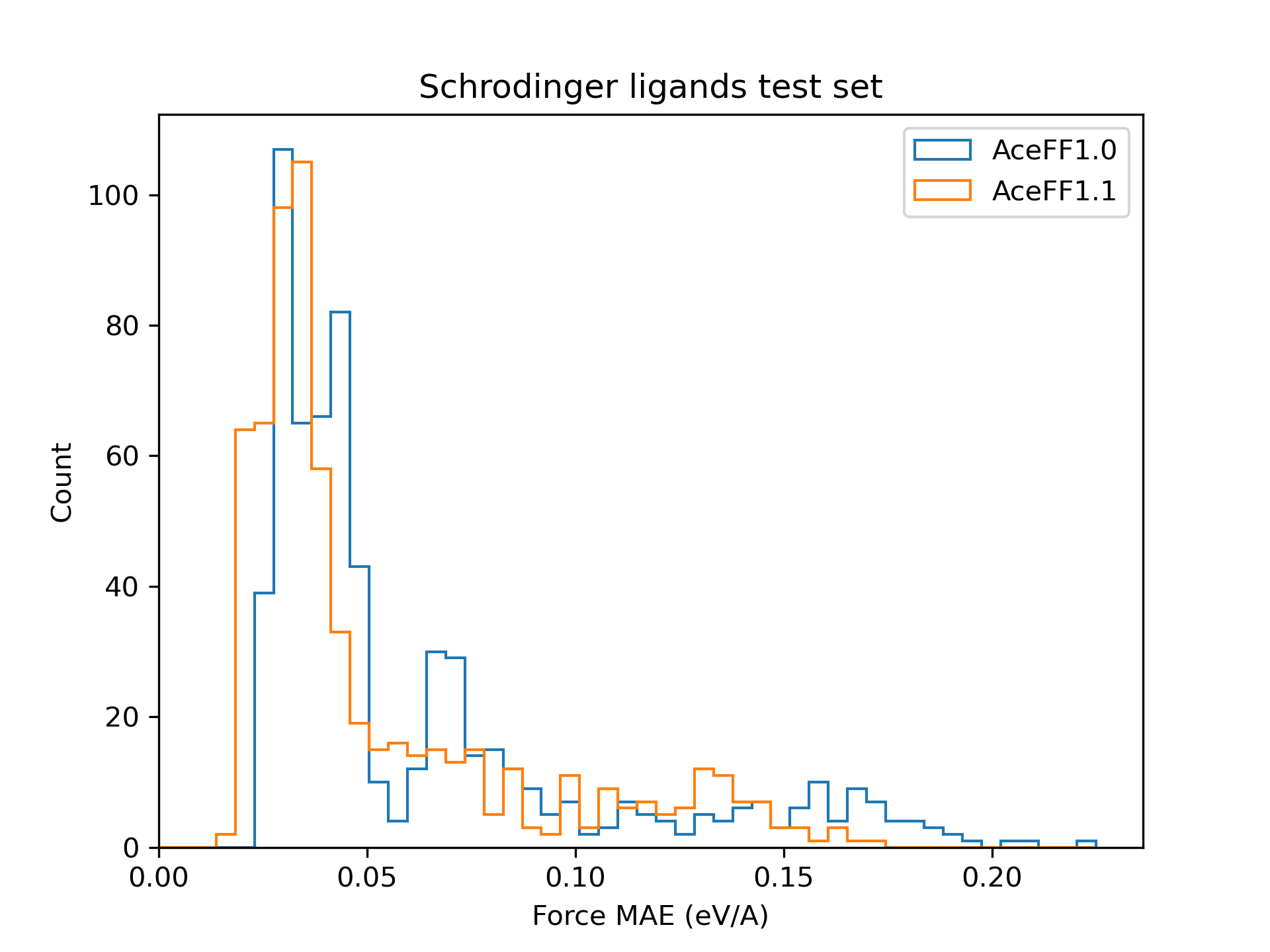
We create our own hold-out test set by labelling 650 ligands from the Schrodinger public binding free energy benchmark (Jacs, Merk, and charge_annhil sets) with AceFF's DFT level of theory. We evaluate the Force MAE of the AceFF predictions.
Example notebooks are available in Google Colab, demonstrating the use of Acellera AceFF with OpenMM and ASE.
Run ML potential molecular simulations of a small molecule using ACEMD with this tutorial, e.g., to minimize.
For a tutorial on running mixed protein-ligand simulations, refer to NNP/MM in ACEMD.
[1] Simeon, Guillem, and Gianni De Fabritiis, Tensornet: Cartesian tensor representations for efficient learning of molecular potentials, Advances in Neural Information Processing Systems 36 (2024), https://arxiv.org/abs/2306.06482
[2] Raul P. Pelaez, Guillem Simeon, Raimondas Galvelis, Antonio Mirarchi, Peter Eastman, Stefan Doerr, Philipp Thölke, Thomas E. Markland, Gianni De Fabritiis, TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations, J. Chem. Theory Comput. 2024, 20, 10, 4076–4087, https://arxiv.org/abs/2402.17660
[3] Francesc Sabanés Zariquiey, Stephen E. Farr, Stefan Doerr, Gianni De Fabritiis, QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials, https://arxiv.org/abs/2501.01811 (2025).